La maladie de Charcot-Marie-Tooth (CMT) est un trouble neurologique héréditaire, ayant diverses formes. Elle entraîne des symptômes moteurs et un déficit sensoriel dès l’enfance. Avec une prise en charge adaptée, il est souvent possible de maintenir une bonne qualité de vie.
Qu’est-ce que la maladie de Charcot-Marie-Tooth ?
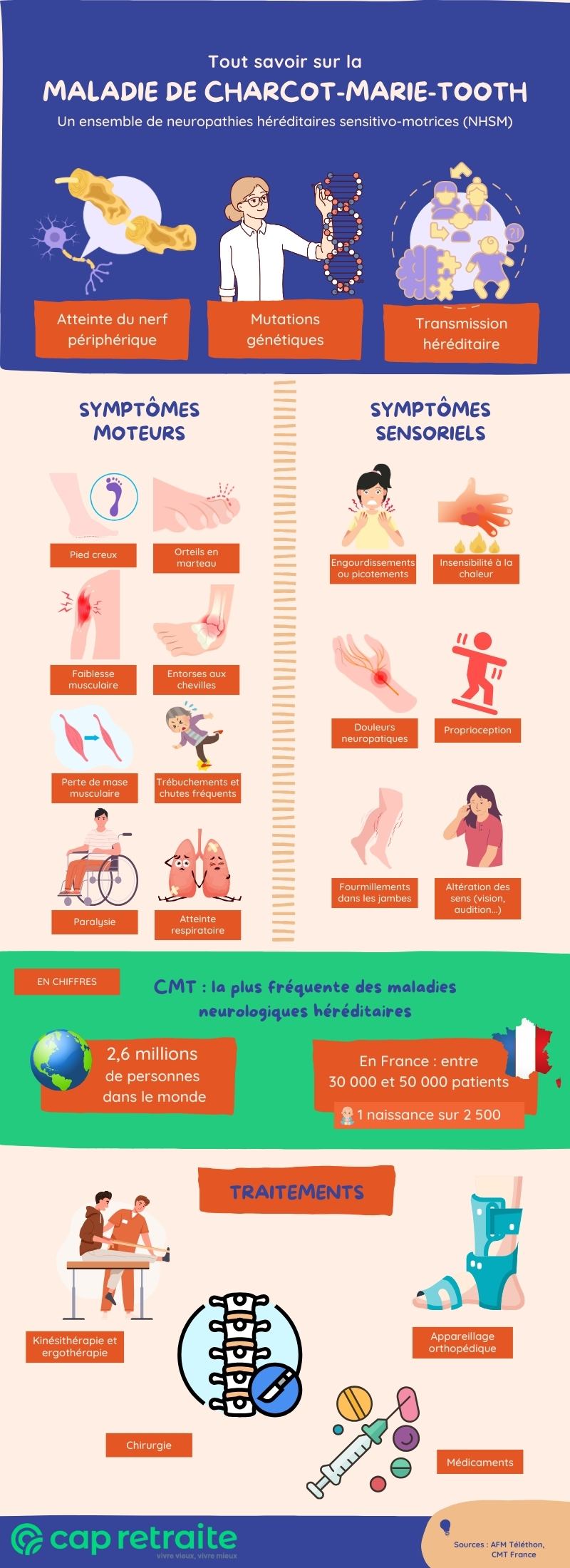
La maladie de Charcot-Marie-Tooth (CMT) fait référence à un ensemble de neuropathies héréditaires sensitivo-motrices (NHSM).
Autrement dit, il s’agit d’une maladie neurologique génétique, qui affecte les nerfs périphériques, entraînant ainsi des difficultés motrices et sensorielles. Elle touche surtout les membres (jambes et bras).
On parle aussi de neuropathie périphérique.
Les nerfs périphériques et la maladie de Charcot-Marie-Tooth
Les nerfs périphériques font la liaison entre le système nerveux central (cerveau et moelle spinale) et le reste du corps. Ils transmettent les signaux nerveux dans les deux sens : du cerveau vers la périphérie (le corps), et inversement.
Ces nerfs contrôlent les muscles et transmettent les informations sensorielles, telles que le sens du toucher, des membres vers le cerveau.
La maladie de Charcot-Marie-Tooth en chiffres
La maladie de Charcot-Marie-Tooth est la plus fréquente des maladies neurologiques héréditaires.
Elle touche quelque 2,6 millions de personnes dans le monde. En France, elle affecte entre 30 000 et 50 000 personnes (soit 1 naissance sur 2 500).
Quels sont les symptômes de la maladie de Charcot-Marie-Tooth ?
Les symptômes de la maladie de Charcot-Marie-Tooth sont les suivants :
- symptômes moteurs :
- faiblesse musculaire, dans les jambes, les chevilles et les pieds, puis dans les mains ;
- perte de masse musculaire ;
- dégradation des compétences de motricité fine, causée par une faiblesse et une atrophie des mains ;
- atrophie des muscles du mollet (mollets de coq) ;
- voûtes plantaires excessivement arquées (pieds creux) ;
- orteils recroquevillés (orteils en marteau) ;
- difficulté à lever le pied en raison d’une incapacité à fléchir la cheville (pied tombant) ;
- démarche maladroite ou levée exagérée du genou (steppage) ;
- difficultés à courir ;
- pertes d’équilibre ;
- chutes ou trébuchements fréquents, en raison de troubles de la posture ;
- entorses fréquentes à la cheville ;
- douleurs dues aux déformations articulaires ;
- courbure anormale de la colonne vertébrale (scoliose) [fréquente dans la maladie de Charcot-Marie-Tooth de type 4C (CMT4C)] ;
- atteinte respiratoire, si les nerfs contrôlant le diaphragme sont impliqués (plus rare dans les formes classiques) ;
- dysplasie de la hanche – malformation se manifestant par des douleurs à la marche ;
- symptômes sensoriels :
- diminution de la sensation dans les jambes et les pieds, puis dans les mains (de la température, des vibrations et de la douleur) ;
- baisse de la sensibilité profonde (proprioception), c’est-à-dire de la perception de la position des parties du corps (sans l’aide de la vue) ;
- douleurs neuropathiques dans les extrémités : sensation de picotement ou de brûlure ;
- altération ou perte d’autres sens, tels que la vision et l’audition (une surdité parfois soudaine peut survenir dans certaines formes de la CMT).
Les symptômes varient d’une forme à l’autre de la maladie et entre les patients eux-mêmes. Dans certains cas, la personne peut ne présenter que des déformations, comme les pieds creux et les orteils en griffes.
Quels sont les premiers symptômes de la maladie de Charcot–Marie-Tooth ?
Les principaux symptômes de la maladie de Charcot-Marie-Tooth apparaissent généralement entre 5 et 15 ans. Toutefois, ils peuvent ne se manifester qu’à l’âge adulte.
Les premiers signes peuvent être difficiles à détecter chez les jeunes enfants. Ils incluent souvent une maladresse inhabituelle et une tendance à être sujet aux accidents.
Comment évolue la maladie de Charcot-Marie-Tooth ?
À mesure que la maladie de Charcot-Marie-Tooth progresse, les symptômes s’aggravent graduellement.
La faiblesse et l’atrophie musculaire, ainsi que la perte de sensibilité, touchant d’abord les pieds et les jambes, peuvent se propager aux mains et aux bras. Une paralysie progressive des membres risque même d’intervenir.
Les troubles de la mobilité et de la posture entraînent peu à peu des douleurs musculaires et articulaires.
L’évolution est généralement lente, mais les symptômes peuvent devenir plus invalidants avec l’âge. L’utilisation d’appareils orthopédiques sera parfois nécessaire pour maintenir la mobilité. Au grand âge, le malade en perte d’autonomie bénéficiera d’une prise en charge adaptée en Ehpad.
Quelles sont les causes de la maladie de Charcot-Marie-Tooth ?
La maladie de Charcot-Marie-Tooth (CMT) est due à des mutations génétiques affectant le développement des nerfs périphériques. Ces mutations perturbent la production de protéines essentielles à diverses fonctions cellulaires.
Atteinte axonale ou démyélinisante
Ces mutations peuvent endommager différentes parties du nerf :
- axone – partie du nerf transmettant les signaux électriques entre le cerveau et les membres ;
- gaine de myéline – enveloppe protectrice autour de l’axone, similaire à l’isolation d’un câble électrique, garantissant une transmission rapide des signaux.
Quand l’axone est affecté, il transmet moins efficacement les signaux. Ainsi, les cellules nerveuses périphériques ne peuvent plus activer les muscles ni relayer les informations sensorielles vers le cerveau et la moelle épinière.
Plus de 80 gènes associés à la CMT ont été identifiés à ce jour, chacun pouvant être lié à une ou plusieurs formes de la maladie.
Les modes de transmission héréditaire de la CMT
Les mutations génétiques de la maladie de Charcot-Marie-Tooth (CMT) sont transmises selon trois modes :
- autosomique dominant : une seule copie du gène muté, héritée de l’un des parents, suffit pour causer la maladie. Chaque enfant d’un parent porteur a 50 % de chances d’hériter de la CMT ;
- autosomique récessif : deux copies du gène muté, une de chaque parent, sont nécessaires pour déclencher la maladie. Chaque enfant a 25 % de chances de la développer ;
- Lié à l’X : la mutation est située sur le chromosome X. Les hommes (XY) héritent de l’X de leur mère et du Y de leur père. Les femmes (XX) héritent d’un X de chaque parent. Le fils d’une mère porteuse a 50 % de chances d’hériter du trouble. Une femme héritant d’un chromosome X touché n’aura que peu ou pas de symptômes, l’autre chromosome X compensant.
Les mutations spontanées « de novo »
Une mutation survient parfois spontanément lors de la conception de l’enfant, sans être transmise par ses parents. Elle peut ensuite être transmise à ses descendants.
Les facteurs de risque de la maladie de Charcot-Marie-Tooth
- hérédité : le principal facteur de risque est d’avoir un parent atteint ou porteur de la maladie ;
- autres neuropathies : des troubles tels que le diabète peuvent causer des symptômes similaires ou aggraver ceux de la CMT ;
- médicaments : certains médicaments, notamment pour la chimiothérapie (vincristine, paclitaxel…), peuvent exacerber les symptômes de la CMT.
La maladie de Charcot-Marie-Tooth est une condition génétique complexe impliquant diverses mutations et différents modes de transmission. La variabilité des gènes impliqués et la manière dont ils affectent les nerfs expliquent la diversité des symptômes et des formes de la maladie.
Quelles sont les différentes formes de la maladie de Charcot-Marie-Tooth ?
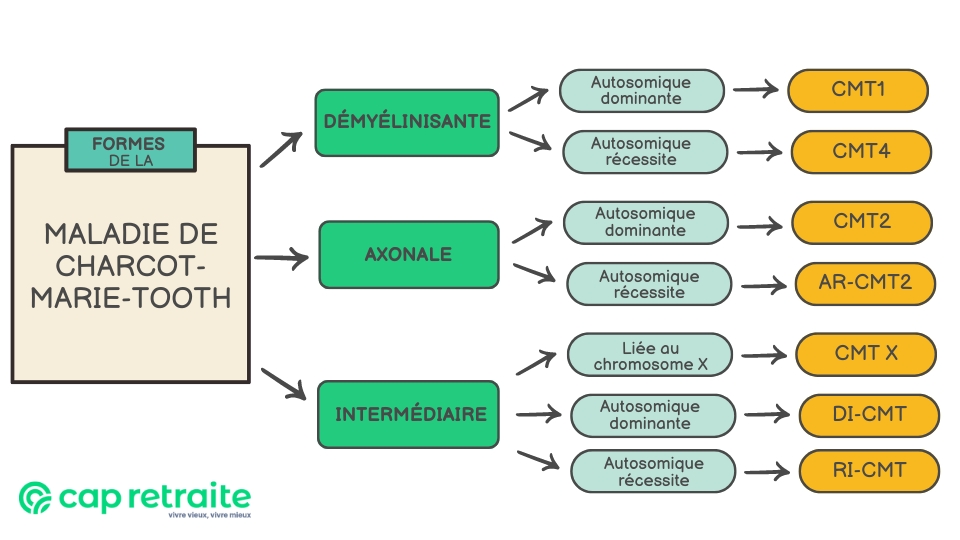
Il existe sept principales formes de la maladie, de Charcot-Marie-Tooth, ensuite réparties en sous-types selon la mutation génétique.
Classification des maladies de Charcot-Marie-Tooth
Les maladies de Charcot-Marie-Tooth sont classées d’après trois critères :
- la partie du nerf périphérique atteinte, déterminée en mesurant les vitesses de conduction nerveuse :
Forme | Vitesse de conduction nerveuse |
|---|---|
CMT axonale | Normale ou quelque peu réduite : supérieure à 38 m/s |
CMT démyélinisante | Réduite : inférieure à 25 m/s |
CMT intermédiaire (mixte) | Intermédiaire : entre 25m/s et 38 m/s |
- le mode de transmission génétique : autosomique dominant, autosomique récessif ou lié à l’X ;
- l’anomalie génétique en cause (la mutation).
Les deux premiers critères définissent les principales formes. Le troisième distingue de nombreux sous-types.
*Il existait autrefois une CMT3. C’est aujourd’hui une neuropathie démyélinisante individualisée, le syndrome de Dejerine-Sottas (SDS).
Quelques exemples de formes de la CMT
- Maladie de Charcot-Marie-Tooth type 1 (CMT1) : due à des anomalies dans la gaine de myéline.
- CMT1A : causée par une duplication du gène PMP22 sur le chromosome 17, entraînant une surproduction de la protéine PMP22, essentielle à la gaine de myéline. Elle se manifeste par une faiblesse musculaire et une atrophie des jambes, des mains, et des pertes sensorielles. C’est la maladie de Charcot-Marie-Tooth la plus fréquente. Elle représente plus de la moitié des cas de CMT.
- CMT1B : résulte de mutations dans le gène MPZ, qui code la protéine zéro de myéline (P0). Plus de 120 mutations ponctuelles ont été identifiées dans ce gène. Les symptômes sont similaires à ceux de la CMT1A.
- Maladie de Charcot-Marie-Tooth type 2 (CMT2) : résulte d’anomalies dans l’axone. Ce trouble autosomique dominant a plus d’une douzaine de sous-types. Certains entraînent une atteinte des cordes vocales et des troubles respiratoires.
- Maladie de Charcot-Marie-Tooth type 4 (CMT4) : comprend plusieurs sous-types de neuropathies démyélinisantes rares. Héritée de manière autosomique récessive, elle affecte des populations ethniques spécifiques. Les symptômes se déclarent souvent par une faiblesse des jambes tôt dans l’enfance, la marche devenant souvent impossible à l’adolescence.
- Maladie de Charcot-Marie-Tooth liée à l’X type 1 (CMTX1) : deuxième forme la plus courante de CMT, elle est liée au chromosome X. Elle touche les garçons plus sévèrement : perte sensorielle, faiblesse et atrophie des muscles distaux des membres, et perte des réflexes musculaires.
Comment est diagnostiquée la CMT ?
Le diagnostic de la maladie de Charcot-Marie-Tooth repose sur une combinaison d’observations cliniques et de tests spécifiques.
Consultation médicale et examen clinique
Le diagnostic commence par une anamnèse détaillée (questions sur les signes observés, ainsi que les antécédents médicaux et familiaux) et un examen neurologique.
Le médecin cherche notamment les signes suivants :
- faiblesse musculaire ;
- diminution de la masse musculaire ;
- réflexes tendineux réduits ;
- pertes sensorielles ;
- déformations des pieds et autres problèmes orthopédiques.
Si le médecin traitant suspecte une CMT, il oriente le patient vers un neurologue pour approfondir les examens.
Tests diagnostiques
Plusieurs tests peuvent être recommandés pour confirmer le diagnostic et déterminer l’étendue des dommages nerveux :
- Études de conduction nerveuse : elles évaluent la qualité des signaux électriques transmis par les nerfs. Ces derniers sont stimulés par des chocs électriques à plusieurs endroits de leur trajet jusqu’au muscle. Le temps écoulé entre la stimulation et la contraction du muscle est alors mesuré. C’est ce qu’on appelle la « vitesse de conduction ». Un signal lent ou faible suggère une neuropathie telle que la CMT. Ces études permettent de distinguer entre les formes axonales et démyélinisantes.
- Électromyographie (EMG) : une fine électrode en forme d’aiguille est insérée dans le muscle pour mesurer son activité électrique, au repos et lors de la contraction. Plusieurs formes de CMT entraînent une modification de l’activité électrique des muscles, pouvant être détectée grâce à un EMG.
- Tests génétiques : des échantillons de sang ou de salive sont analysés pour déterminer les mutations génétiques courantes responsables de la CMT.
- Biopsie nerveuse : une petite portion d’un nerf périphérique est prélevée pour analyse en laboratoire, afin de distinguer la CMT des autres neuropathies.
Peut-on guérir de la maladie de Charcot-Marie-Tooth ?
Il n’existe pas de traitement curatif de la maladie de Charcot-Marie-Tooth. Autrement dit, il n’est pas possible d’en interrompre l’évolution ni de l’inverser.
Il existe néanmoins des traitements symptomatiques et orthopédiques pour améliorer la qualité de vie du patient et l’aider à préserver un maximum d’autonomie.
Rééducation physique
La kinésithérapie et l’ergothérapie aident à maintenir au maximum la mobilité, la flexibilité et la force musculaire.
Un programme d’étirements et d’exercices adaptés et réguliers peut prévenir ou retarder la dégénérescence nerveuse et la faiblesse musculaire. Une activité physique modérée est aussi souvent bénéfique (natation, gymnastique, etc.)
Appareillage orthopédique
Des appareils orthopédiques fournissent un soutien supplémentaire et améliorent la marche :
- orthèses plantaires pour les pieds creux,
- attelles pour les chevilles et les jambes,
- chaussures sur mesure,
- semelles adaptées.
Les orthèses pour les pouces permettent de renforcer la préhension et les mouvements fins des mains.
Prise en charge médicamenteuse de la CMT
Des médicaments sont parfois prescrits pour soulager les douleurs musculaires ou neuropathiques :
- anti-inflammatoires non stéroïdiens (AINS) pour les douleurs articulaires et musculaires,
- antidépresseurs tricycliques ou anticonvulsivants pour la douleur neuropathique.
Chirurgie orthopédique pour les cas plus graves
Une intervention chirurgicale peut être nécessaire pour corriger :
- les déformations sévères des extrémités (pieds creux, doigts ou orteils en griffe), de la colonne vertébrale (scoliose) ou d’autres articulations ;
- le steppage dérangeant la marche.
Les techniques chirurgicales existantes sont notamment les suivantes :
- ostéotomie : visant à corriger la forme des os. Elle consiste à couper et repositionner les os pour améliorer l’alignement et la fonction des pieds ;
- arthrodèse : consistant à fusionner plusieurs os d’une articulation pour la stabiliser et la renforcer.
Quelle est la différence entre la maladie de Charcot et la maladie de Charcot-Marie-Tooth ?
La principale différence entre la maladie de Charcot (SLA) et la maladie de Charcot-Marie-Tooth réside dans les systèmes corporels qu’elles affectent.
Ces atteintes distinctes entraînent des symptômes et une prise en charge différents.
Caractéristique | Maladie de Charcot-Marie-Tooth (CMT) | Maladie de Charcot (sclérose latérale amyotrophique – SLA) |
|---|---|---|
Système affecté | Nerfs périphériques | Neurones moteurs du système nerveux central |
Symptômes principaux |
|
|
Cause | Mutations génétiques affectant les nerfs périphériques | Dégénérescence des neurones moteurs |
Mode de transmission | Héréditaire, parfois spontanée | Majoritairement sporadique, parfois héréditaire |
Évolution |
|
|
Traitement | Symptomatique : appareillage, physiothérapie | Symptomatique : médicaments pour ralentir la progression, thérapies de soutien |
Espérance de vie | Normale | Réduite, souvent de 3 à 5 ans après le diagnostic |
Questions fréquentes
Quelle est l’espérance de vie avec la maladie de Charcot-Marie-Tooth ?
La maladie de Charcot-Marie-Tooth n’affecte généralement pas l’espérance de vie. La plupart des patients auront une durée de vie normale.
Toutefois, certaines formes rares de CMT peuvent avoir un pronostic moins favorable. La durée de vie du patient risque d’être réduite par une atteinte respiratoire, l’inactivité, la dépression ou des comorbidités.
Quelles sont les principales complications de la maladie de Charcot-Marie-Tooth ?
Les principales complications de la CMT sont les suivantes :
- blessures et fractures dues à une chute ;
- aggravation des symptômes à cause de certains médicaments ;
- blessures et infections des pieds passant inaperçues, le patient ne ressentant pas la douleur ou la température.
Qui a découvert la maladie de Charcot-Marie-Tooth ?
La CMT a été découverte et décrite à la fin du XIXe siècle par deux neurologues français, Jean-Martin Charcot et Pierre Marie, et leur homologue britannique, Howard Henry Tooth.
Cet article vous a-t-il été utile ?
Notez cet article afin de nous permettre d’améliorer nos contenus.

Réagissez, posez une question…